May 2024
·
21 Reads
Communications Biology


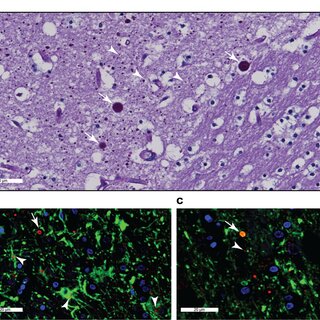
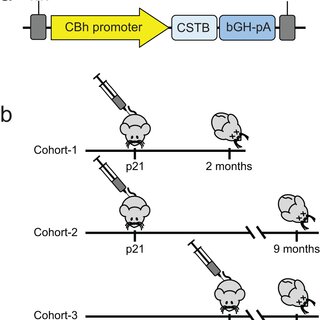
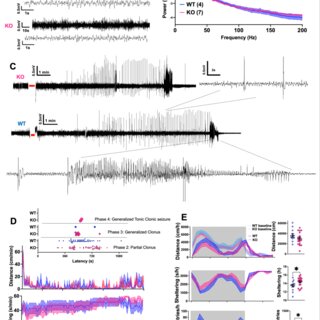
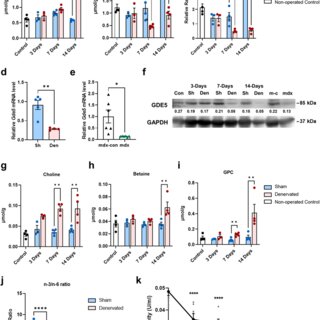
Glycerophosphocholine (GPC) is an important precursor for intracellular choline supply in phosphatidylcholine (PC) metabolism. GDE5/Gpcpd1 hydrolyzes GPC into choline and glycerol 3-phosphate; this study aimed to elucidate its physiological function in vivo. Heterozygous whole-body GDE5-deficient mice reveal a significant GPC accumulation across tissues, while homozygous whole-body knockout results in embryonic lethality. Skeletal muscle-specific GDE5 deletion (Gde5 skKO) exhibits reduced passive force and improved fatigue resistance in electrically stimulated gastrocnemius muscles in vivo. GDE5 deficiency also results in higher glycolytic metabolites and glycogen levels, and glycerophospholipids alteration, including reduced levels of phospholipids that bind polyunsaturated fatty acids (PUFAs), such as DHA. Interestingly, this PC fatty acid compositional change is similar to that observed in skeletal muscles of denervated and Duchenne muscular dystrophy mouse models. These are accompanied by decrease of GDE5 expression, suggesting a regulatory role of GDE5 activity for glycerophospholipid profiles. Furthermore, a DHA-rich diet enhances contractile force and lowers fatigue resistance, suggesting a functional relationship between PC fatty acid composition and muscle function. Finally, skinned fiber experiments show that GDE5 loss increases the probability of the ryanodine receptor opening and lowers the maximum Ca²⁺-activated force. Collectively, GDE5 activity plays roles in PC and glucose/glycogen metabolism in skeletal muscle.